Where allele structure directly impacts clinical interpretation, long-read sequencing provides critical resolution. Clinical PGx value today is concentrated in a defined set of genes with well-established roles in drug metabolism and response.
While many of these genes can be adequately resolved with short-read sequencing, structurally complex loci, such as CYP2D6, HLA, UGT1A1, and select DPYD/TPMT contexts, require long-read approaches to accurately capture duplications, hybrid alleles, and phased variants that determine functional output.
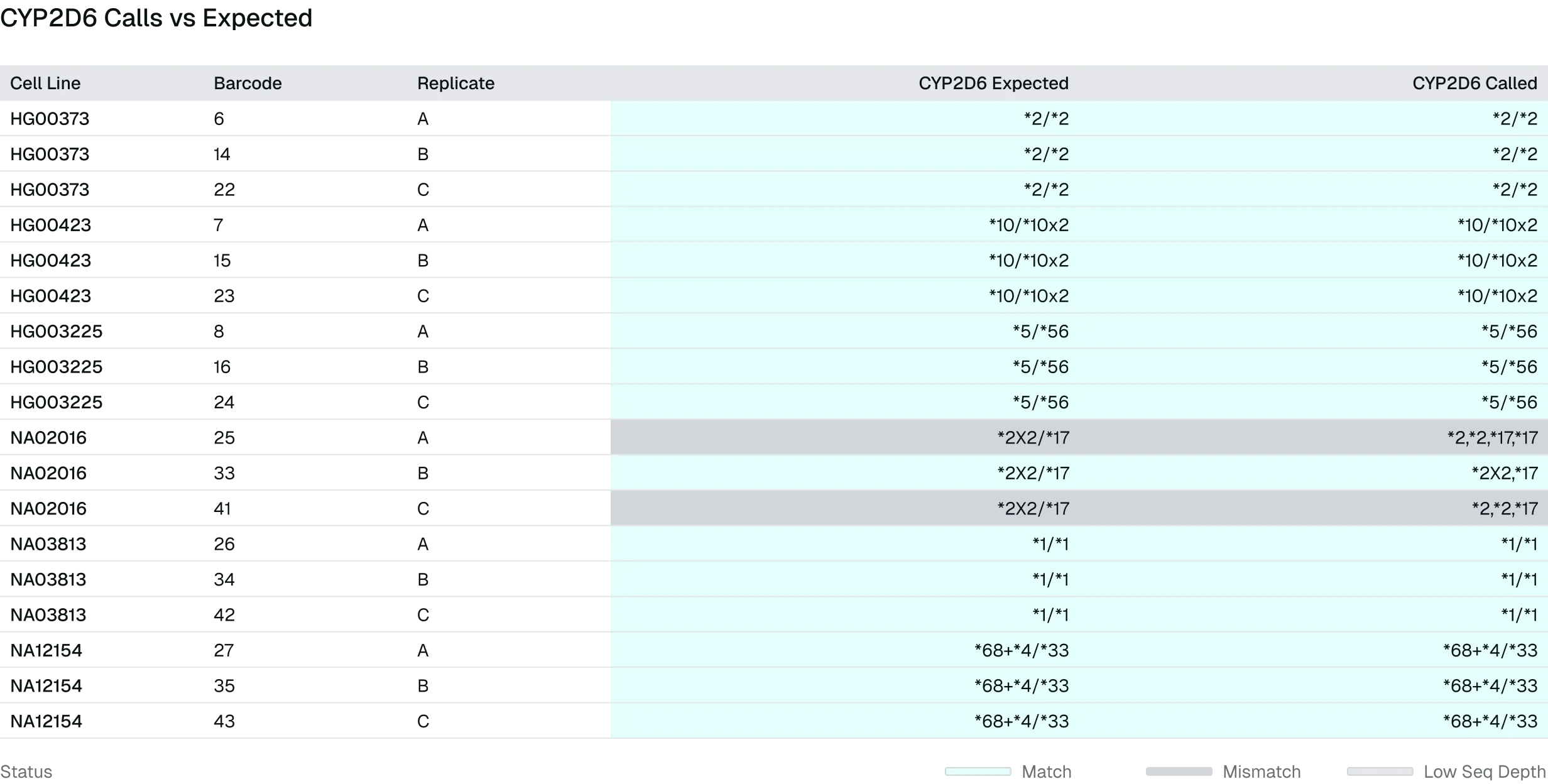
Figure 1. Long-read sequencing enables decision-grade CYP2D6 haplotype and structural variant resolution. Per-sample CYP2D6 star-allele calls, including copy number and hybrid structures (e.g., *1/*4x2, *36+*10), were compared to expected haplotypes across truth-set samples (GIAB, Corielle; ≥30x). Despite minor discrepancies in biologically complex regions, high concordance was observed, demonstrating the ability of long-read sequencing to accurately resolve PGx haplotypes and structural variation in clinically relevant genes.
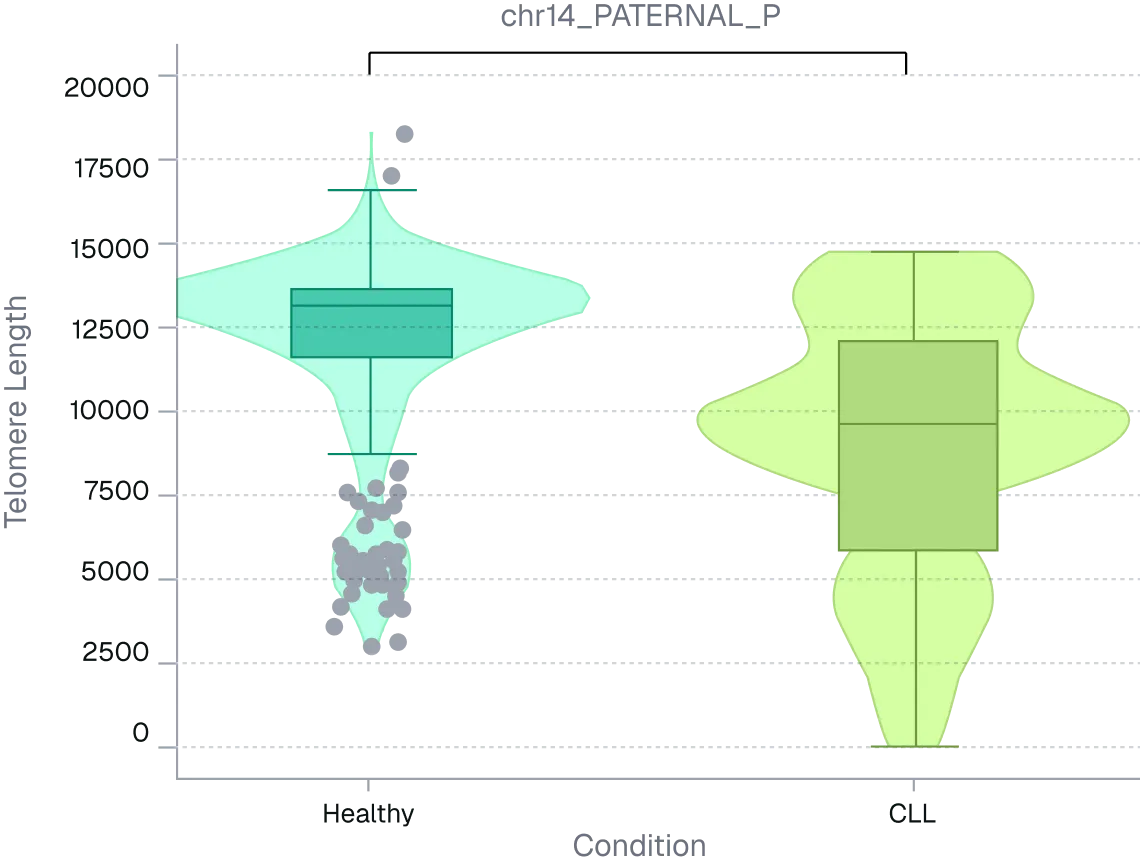
Legacy telomere methods force trade-offs between accessibility and resolution. Techniques such as qPCR, Flow-FISH, and TRF provide global estimates of telomere length but rely on averaged signals across chromosomes, obscuring arm-specific variation, subtelomeric structure, and genomic context.
This lack of resolution limits both research and clinical insight. For researchers, it masks locus-specific mechanisms driving genomic instability, aging biology, and disease progression. For clinicians, it can obscure high-risk profiles, where critically short telomeres on specific chromosome arms remain hidden within a “normal” global average, limiting accurate risk assessment and patient stratification. Long-read sequencing overcomes these limitations by enabling direct, chromosome-specific measurement of native telomere length while preserving subtelomeric context.
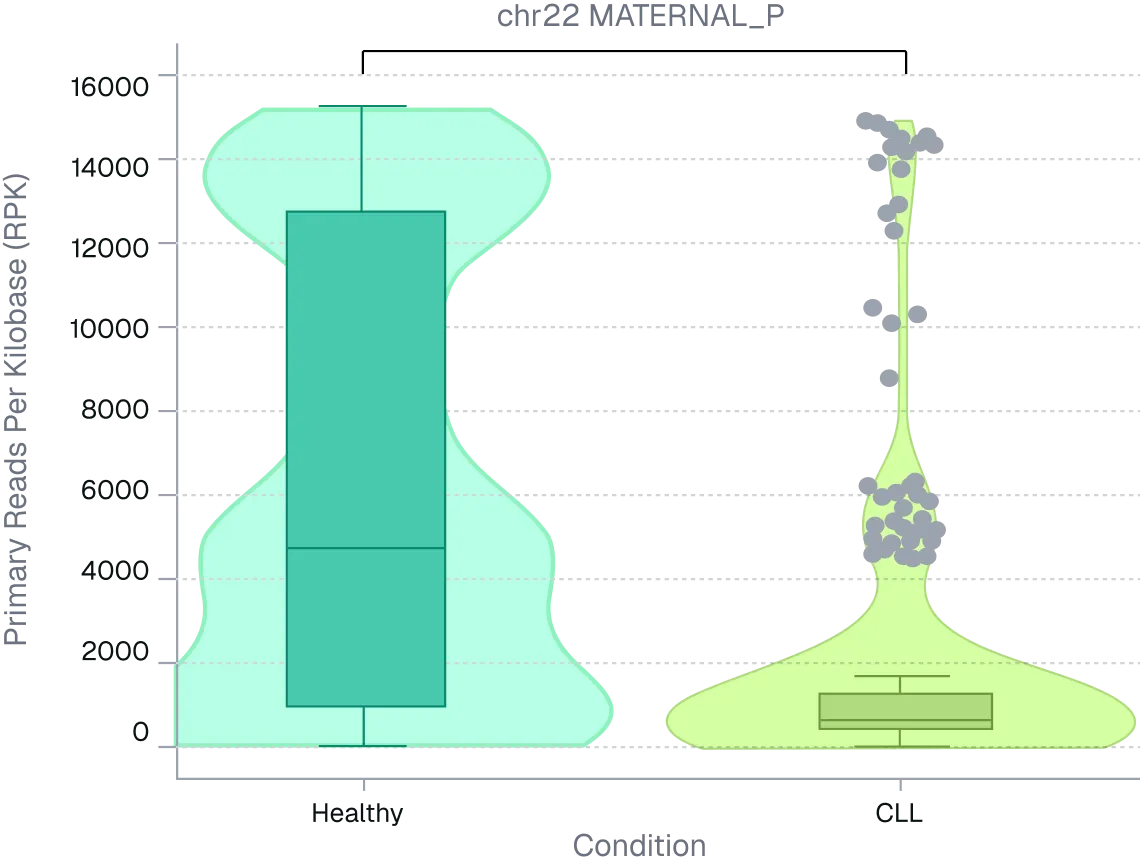
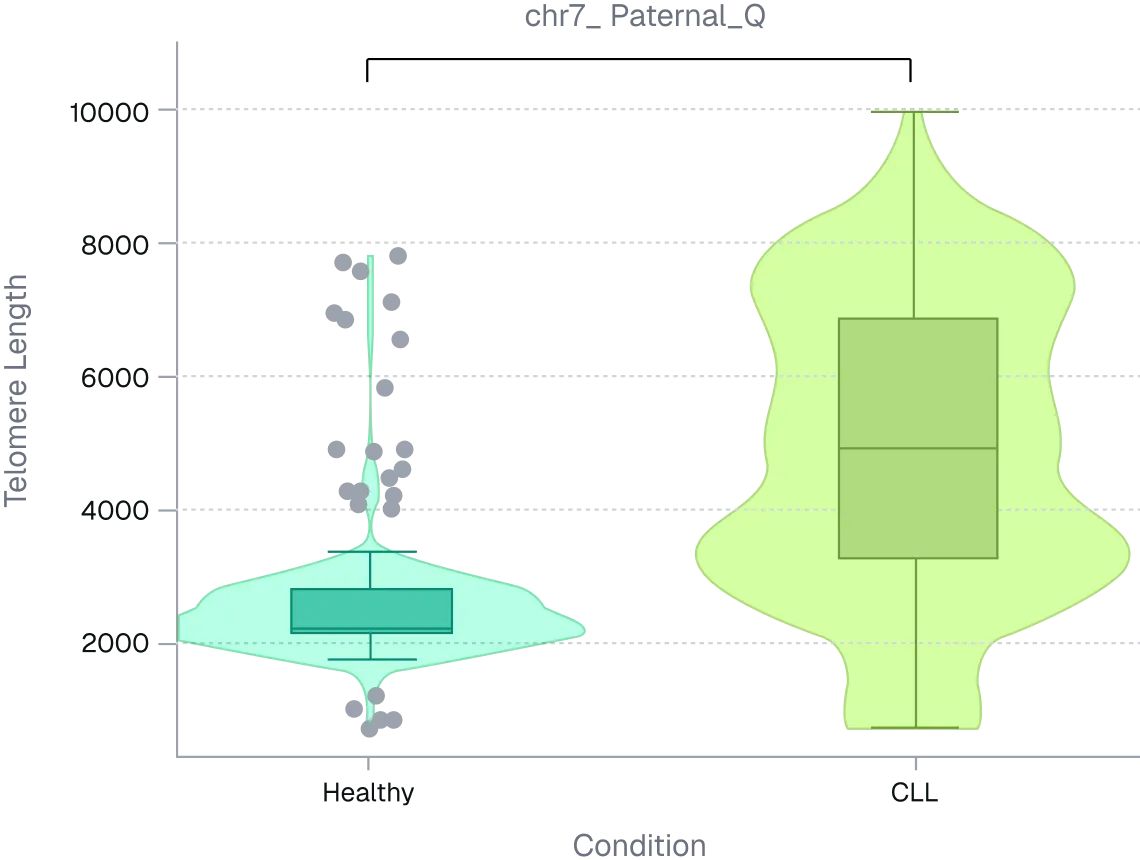
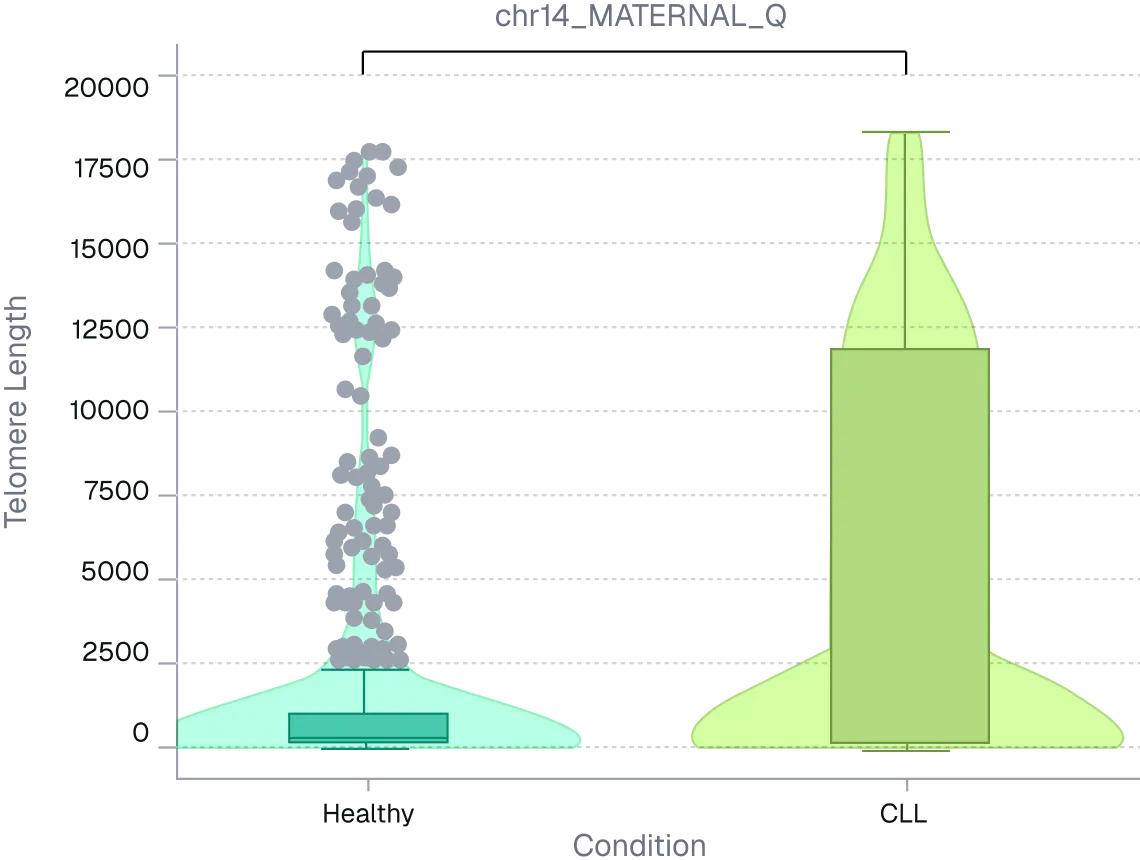
Figure 2. ONT long-read sequencing resolves telomere arm-length differences masked by global averages. Per-arm telomere length distributions were compared between healthy and CLL PBMC donors using long-read sequencing. Despite modest global shifts, multiple chromosome arms showed significant median differences (~2.5–5.5 kb).
Clinically-relevant genes with highly homologous pseudogenes represent a major blind spot for short-read sequencing, where high sequence similarity leads to misalignment, off-target mapping, and unreliable variant calls. This challenge is exemplified by PKD1 (Polycystic Kidney Disease 1), which shares >97% homology with multiple pseudogenes, making accurate variant detection difficult with short reads.
Long-read sequencing overcomes this limitation by spanning homologous regions within single molecules, enabling accurate mapping and variant resolution without reliance on complex PCR strategies. In targeted nanopore workflows, this also allows simultaneous interrogation of sequence variation and methylation within structurally complex loci.

Figure 3. ONT long-read sequencing enables accurate alignment in highly homologous PKD1 pseudogenes. Read alignments across PKD1 and its pseudogenes (>97% homology) demonstrate accurate mapping to the functional gene, with significantly higher reads per kilobase compared to homologous loci. Across 634 target regions, SNV detection achieved 98.5% precision and 99.5% recall with >120× mean coverage and >1,600x enrichment.
Novel and non-model genomes often present challenges that limit accurate assembly with short-read sequencing, including high repetitive content, structural complexity, and degraded or low-quality input DNA. These constraints frequently result in fragmented assemblies, misaligned contigs, and incomplete representation of functionally important regions.
Long-read sequencing overcomes these limitations by spanning repetitive elements and preserving long-range genomic context, enabling more contiguous and complete assemblies. Wasatch BioLabs applies optimized extraction, high-molecular-weight DNA workflows, and long-read nanopore sequencing to generate high-quality genome assemblies from challenging samples, supporting downstream functional, evolutionary, and translational research.
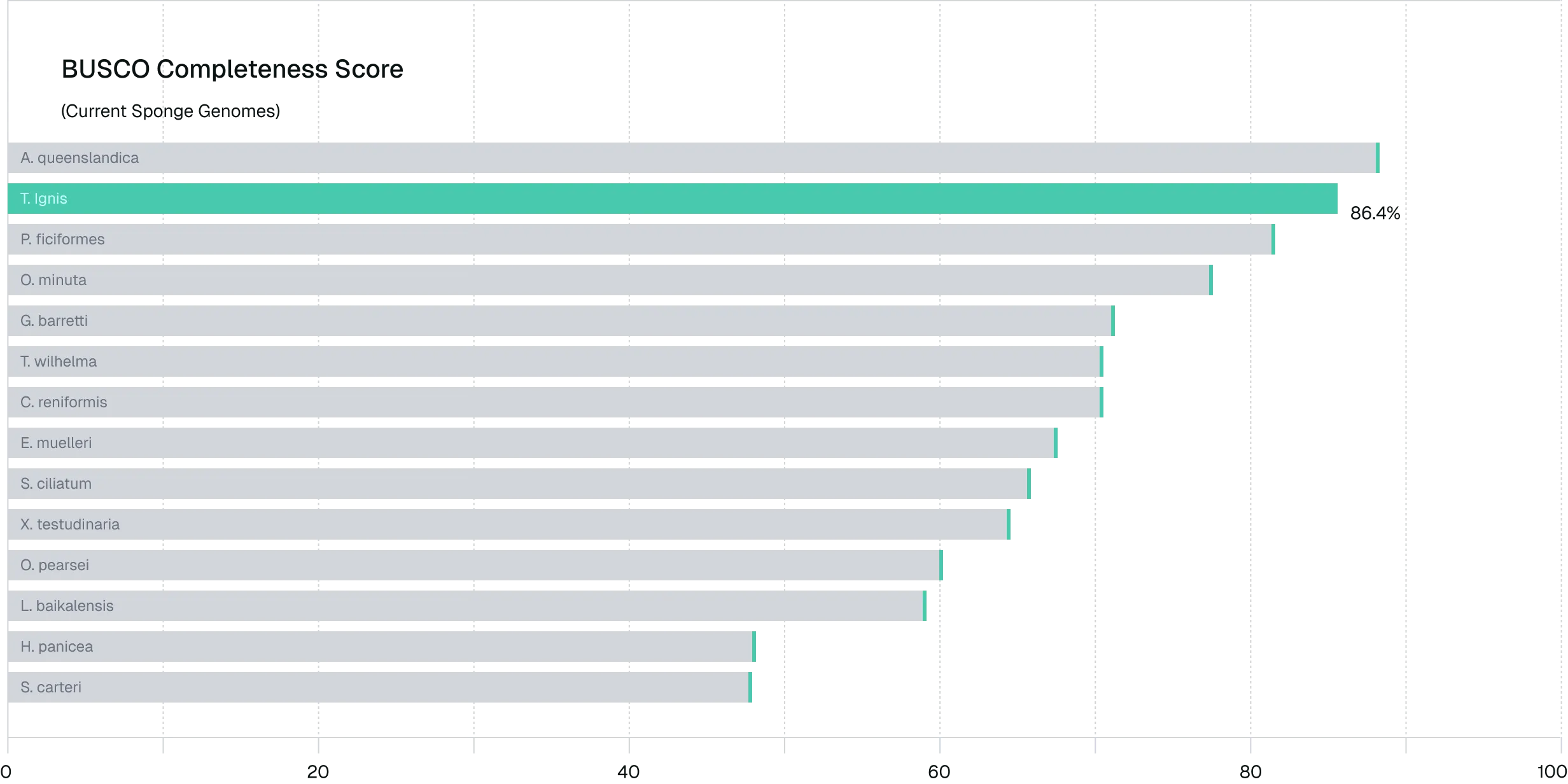
Figure 4. Long-read sequencing enables high-quality, functionally-complete de novo genome assembly. BUSCO analysis of the Tedania ignis genome assembly demonstrates high completeness (86.4%), indicating strong representation of conserved orthologs relative to reference species. RNA-seq validation showed >82% alignment to the assembled genome, supporting structural accuracy and functional integrity, and highlighting the effectiveness of long-read sequencing for complex, non-model genome assembly.
Accurate microbial community profiling requires resolving species-level composition without distortion from amplification bias or incomplete genomic context. Both conventional and long-read targeted 16S approaches rely on PCR and other enrichment methods, introducing bias and artifacts while excluding broader genomic and epigenetic information.
Long-read, whole-genome sequencing overcomes these limitations by directly profiling native DNA, enabling species- and strain-level resolution with integrated methylation detection. Wasatch BioLabs’ Direct Whole Methylome Sequencing (dWMS) platform delivers a PCR-free, scalable workflow that preserves native genomic and epigenetic context, supporting high-fidelity microbial profiling even at reduced sequencing depth.
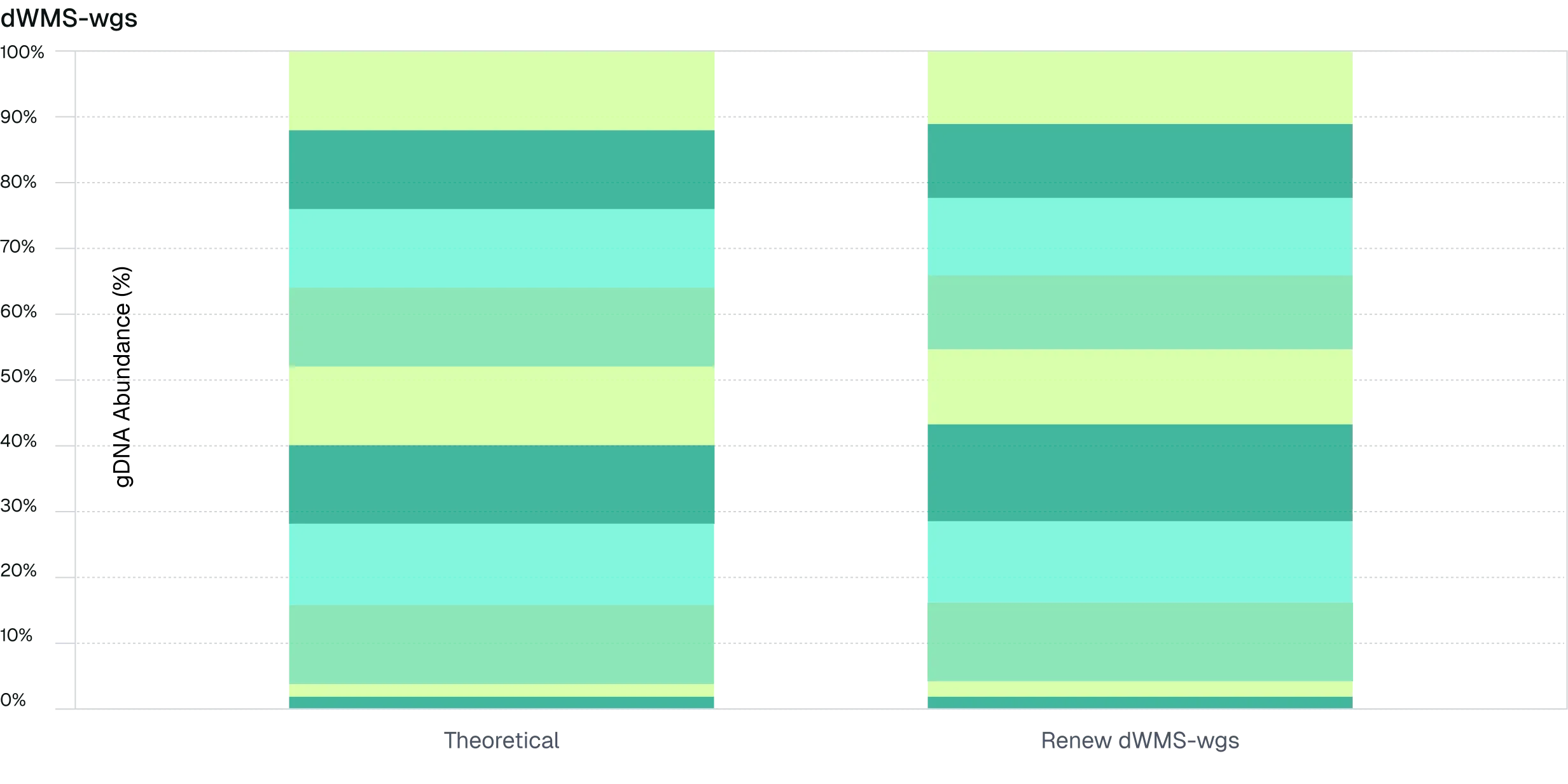
Figure 5. dWMS accurately recapitulates microbial community composition and outperforms targeted 16S methods. Whole-genome dWMS sequencing of a 10-species microbial standard achieved accurate species identification and relative abundance estimates, with an average deviation of 5.7% from theoretical composition. Compared to both Illumina short-read and ONT long-read 16S workflows, dWMS demonstrated improved species-level accuracy and reduced bias, maintaining performance under downsampled conditions and highlighting the value of long-read, PCR-free profiling.